Reaching the 800th article on this website is a remarkable achievement.
It has taken 10 years since the website was launched. There is a veritable encyclopaedia of aHUS articles. An A to Z of aHUS.
If we had to rely on published journals we would not likely have have more than ten articles now out in the public domain.
Global Action is thwarted when it tries to publish material that way. That is life of the patient researcher!
The most recent attempt was trying to get an anniversary article published in the journal where the term HUSs appeared for the first time.
It was also a way of getting the English translation version of that article into the public domain.
The original article was in German and other than its title it cannot be accessed through any of the important clinical research articles repositories , PubMed etc. It is not even showing in the original publication’s archives.
For a time that publication was supportive of an “opinion” style article from Global Action with the translation as an Appendix and it looked a possibility and then no more was heard with no explanation despite our asking.
That is how authors are treated. It may well be that von Gasser’s article from 70 years ago would not have passed the ethical requirements expected these days. But at least that would be a reason to be turned down.
The time and effort spent on it is not entirely wasted as this website can now benefit and mark its 800th blog article with the opinion piece about the article which started HUSs off on its journey to today.
So here it is :
On the 70th anniversary of Conrad von Gasser’s seminal article on the hemolytic uremic syndromes publication in the Swiss Medical Weekly
Len Woodward a
a aHUS alliance Global Action , Cheshire, UK
Today the term atypical hemolytic uremic syndrome (aHUS) embraces a range of thrombotic microangiopathic conditions or TMAs.1 It is defined by the presence of the triad of medical conditions of hemolytic anaemia, thrombocytopenia and kidney injury.2 This triad was first described by Conrad von Gasser, a pediatric hematologist at the Kinderspital, Zurich. He had led a group of researchers which also included pathologists. In 1954 the research team begun to focus on the clinical records and postmortem pathological findings for five young children who had been admitted to the Kinderspital during the previous two years. Each child had presented differently but all appeared to have low red blood cells low platelets and kidney impairment. All had died. During the research, Gasser coined the term Hemolytic Uremic Syndromes which he typed on one child’s death certificate.3 An article about the syndromes subsequently appeared in the Swiss Medical Weekly on 20 September 1955.4
In subsequent decades it would be rare for the Gasser et al article about the HUSs not to be cited as a reference to define the diseases. In time the HUSs developed into two categories. Hemolytic Uremic Syndrome became linked to Shiga Toxin producing E. Coli, a specific infection trigger of TMA5 ; and aHUS was adopted for other TMA triggers, including uncontrolled overactivation of the alternative pathway of the complement system.6 In 1955 Von Gasser may have been familiar with Bacterium coli commune7but not aware of a specific involvement of a STEC infection or complement system genetics8 in his HUSs, but he had noted that “strong antigen antibody reactions occurred in these children following the severe and mild torpid infections” ( in the article below).
When aHUS alliance Global Action, an incorporated charity advocating for aHUS patients9 began writing a history of aHUS it sought a copy of the original Von Gasser article. Written in an era when articles were distributed to subscribers, libraries and institutions all that could be hoped for was that a digitised version had been made. It was not known whether the original article had been digitised or translated into English. All that could be found online was the article title and citation reference on PubMed with no link to a full text or an abstract.4 Similarly with the reprise of the article in a “Wissenschaftliche Rosinen “ series by Swiss Medical Weekly in 1995.9.
Eventually a pdf print of the original article was traced. The charity commissioned an English translation of the original article which had been written in German.
In honour of the first HUSs research cohort10 and on the seventieth anniversary of its first publication in Swiss Medical Weekly,4, an English language version of Professor Conrad von Gasser’s seminal work on the hemolytic uremic syndromes is presented.
References :
1 . Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy:
conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney
Int. 2017;91(3):539-551. doi:10.1016/j.kint.2016.10.005
- Loirat, C., Frémeaux-Bacchi, V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 6, 60 (2011).
https://doi.org/10.1186/1750-1172-6-60 - Gautier C., Siebenmann, Chapter1, The Birth of the Hemolytic Uremic Syndrome, Kaplan B.S., Trompeter
R.S., and Moake J.L., Eds, Hemolytic Uremic Syndrome and Thrombotic Thrombocytopenic Purpura, Marcel
Dekker, Inc. New York, NY, 1992 - GASSER C, GAUTIER E, STECK A, SIEBENMANN RE, OECHSLIN R. Hämolytisch-urämische Syndrome: bilaterale
Nierenrindennekrosen bei akuten erworbenen hämolytischen Anämien [Hemolytic-uremic syndrome: bilateral
necrosis of the renal cortex in acute acquired hemolytic anemia]. Schweiz Med Wochenschr. 1955;85(38-
39):905-909. - Jokiranta TS. HUS and atypical HUS. Blood. 2017 May 25;129(21):2847-2856. doi: 10.1182/blood-2016-11-
- Epub 2017 Apr 17. PMID: 28416508; PMCID: PMC5445567.
- Afshar-Kharghan V. Atypical hemolytic uremic syndrome. Hematology Am Soc Hematol Educ Program. 2016
Dec 2;2016(1):217-225. doi: 10.1182/asheducation-2016.1.217. PMID: 27913483; PMCID: PMC6142509. - Etymologia: Escherichia coli. Emerging Infectious Diseases. 2015;21(8):1310. doi:10.3201/eid2108.et2108.
- Paul Warwicker, Timothy H.J. Goodship et al, Genetic studies into inherited and sporadic hemolytic uremic
syndrome, Kidney International, Volume 53, Issue 4,1998,Pages 836-844,ISSN 0085-2538,
https://doi.org/10.1111/j.1523-1755.1998.00824.x. - Gasser C, Gautier E, Steck A, Siebenmann RE, Oechslin R. Wissenschaftliche Rosinen aus 125 Jahren SMW.
Hämolytisch-urämische Syndrome: bilaterale Nierenrindennekrosen bei akuten erworbenen hämolytischen
Anämien. 1925 [Scientific raisins from 127 years SMW (Swiss Medical Weekly). Hemolytic-uremic syndrome:
bilateral kidney cortex necrosis in acute acquired hemolytic anemia. 1925]. Schweiz Med Wochenschr.
1995;125(51-52):2528-2532.
Acknowledgement : all patients to day are grateful to the five patients whose deaths inspired the research which created the name that is shared with them today.
APPENDIX A
THE ORIGINAL ARTICLE
From the University Paediatric Hospital (Director: Prof. G. Fanconi) and the Institute of Pathology and Anatomy (Director: Prof. E. Uehlinger), Zurich
Haemolytic-uraemic syndromes:
Bilateral renal cortical necrosis in acute acquired
haemolytic anaemias
By C. Gasser, E. Gautier and Annemarie Steck (clinical section) and R.E. Siebenmann and R. Oechslin (anatomic pathology section)
From a group of 10 fatal cases associated with uraemia and haemolytic anaemia, five acute cases are described, which are characterised by the sudden onset of acute intravascular haemolysis of unknown cause in conjunction with acute renal failure, which from a clinical, and particularly from a pathological and anatomical perspective, does not correspond to haemolytic-uraemic or crush syndrome. As our cases are characterised, in addition to haemolytic anaemia, by thrombocytopenic purpura, they are reminiscent of thrombotic-thrombocytopenic purpura as described by Moschcowitz and Singer and of thrombotic microangiopathy as described by Symmers and finally of the syndrome “thrombocytopenic purpura with acquired haemolytic anaemia” described by Evans. However, severe renal failure is generally absent in these diseases.
Case 1. W. Susi, DOB 2 June 1946, J. No. 831/53. A seven-year-old girl previously always in good health apart from hives as a reaction to strawberries, falls ill within a few hours with acute acquired haemolytic anaemia with severe haemoglobinuria, icterus, haemorrhagic diathesis and mild oedema. Four days later the icterus disappears and there is a slight increase in anaemia. Despite an apparent improvement in her condition, the child dies three days later from the consequences of severe renal failure.
The precise analysis of this case (Fig. 1) indicates: erythrocyte damage at onset with decreased osmotic resistance, no evidence of autoantibodies; no erythropoietic regeneration. Ecchymosis with moderate thrombocytopaenia and significantly increased antithrombin levels. On admission, we also find renal symptoms: mild oedema – particularly of the eyelids, elevated blood pressure, oliguria, high specific gravity, albuminuria, haematuria and cylindruria, i.e. symptoms that are not all consistent with the initial stage of crush syndrome but are consistent with acute glomerulonephritis. The elevated BUN and potassium levels are in part the consequence of severe haemolysis.
With ACTH treatment the intravascular haemolysis stops and a strong erythropoietic regeneration begins. However, the increased blood degradation persists and even a whole-blood transfusion is unable to increase the haemoglobin, which indicates a haemolytically active, blood group-non-specific factor in the plasma. The second half of the disease duration is dominated by increasingly prominent renal function failure, oliguria, continuous increase in BUN levels, metabolic acidosis. Hypochloraemia is probably a consequence of severe vomiting; sodium levels are normal. On the eighth day the previously normal sodium levels increase and at the same time symptoms of acute heart failure appear; an ECG indicates typical manifestations of hyperkalaemia. The exchange transfusion performed in extremis is unable to prevent the fatal outcome. Death on the eighth day.
The autopsy (p. no. 769/53) indicates neither haemolytic-uraemic/crush syndrome nor glomerulonephritis, but it does indicate extensive acute glomerular necrosis with isolated tubular necrotic lesions as a specific form of bilateral renal cortical necrosis. Apart from moderate haemoglobinuria, the only indication of crush syndrome is provided by isolated lesions characteristic of tubular necrosis (Fig.3). The consequences of uraemia are epithelial hyperplasia and eosinophilia of the adenohypophysis. Eccentric hypertrophy of both ventricles, splenic atherosclerosis and acute organ congestion with cavity effusion indicate decompensated renal hypertension. The haemorrhagic diathesis is manifested by mucosal bleeding and a minor pontine haemorrhage.
The autopsy (p. no. 769/53) indicates neither haemolytic-uraemic/crush syndrome nor glomerulonephritis, but it does indicate extensive acute glomerular necrosis with isolated tubular necrotic lesions as a specific form of bilateral renal cortical necrosis. Apart from moderate haemoglobinuria, the only indication of crush syndrome is provided by isolated lesions characteristic of tubular necrosis (Fig.3). The consequences of uraemia are epithelial hyperplasia and eosinophilia of the adenohypophysis. Eccentric hypertrophy of both ventricles, splenic atherosclerosis and acute organ congestion with cavity effusion indicate decompensated renal hypertension. The haemorrhagic diathesis is manifested by mucosal bleeding and a minor pontine haemorrhage.

Fig 1.* Course curve case 1
.
Case 2. S. Carmen, DOB 11 December 1953, J. No. 2815/54. Two-month-old baby with purulent rhinitis. Treatment with sulfonamide ointments. Two days before hospitalisation: occurrence of brownish urine, diarrhoea, rapidly increasing pallor, spasms and clouded consciousness. On admission, severe anaemia (26% Hb) with haemoglobinaemia. Morphologically severe erythrocyte decay. No autoantibodies, no evidence of erythropoiesis. Elevated erythropoiesis in the bone marrow. Thrombocytopaenia. Increasing susceptibility to spasms. On the third day, transition to complete anuria with appearance of oedema, high BUN and phosphorus levels. Rapid deterioration. An unsuccessful attempt is made to give the dying child an exchange transfusion. Death on the fourth day. Disease progression contradicts the initially assumed diagnosis of drug-induced toxic-haemolytic anaemia.
The autopsy (p. no. 229/54) again indicates bilateral renal cortical necrosis in addition to pronounced haematuria and haemoglobinuria. Not only are these cortical necrotic lesions predominantly haemorrhagic, but there is also extensive mucosal bleeding, subserosal bleeding and bone marrow and subarachnoid haemorrhage indicative of haemorrhagic diathesis. Erythroid hyperplasia is present in the bone marrow. Abundant haemosiderin depositions are present in the spleen, liver and bone marrow.
Case 3. H. Barbara, DOB 29 December 1953, J. No. 4093/54. Seven-month-old girl, already pale, suffering from acute diarrhoea and vomiting. Three days later, admission with pale-yellow colour, macroscopic haemoglobinaemia and haemoglobinuria. Symptoms of severe renal failure appear three weeks later, i.e. a week before death.
Disease progression (Fig. 2) is characterised by acute acquired haemolytic anaemia with erythrocyte decay and significantly reduced osmotic resistance, elevated bilirubin and high serum iron level, but no autoantibodies. Start of sustained regeneration soon afterwards. Despite thrombocytopaenia no haemorrhagic diathesis. The electrolyte balance is still almost normal at onset. Stool culture negative. Treatment with antibiotics, ACTH and blood transfusions. In the third week increasing oedema, reduced urine output, further vomiting, increased BUN and phosphorus levels, metabolic acidosis with hypochloraemia, normal sodium as well as increasing hypoproteinaemia. At the start of the fourth week, occurrence of haemorrhagic diatheses and hemiparesis. An exchange transfusion results in no significant improvement in the progression of haemolytic anaemia or in renal function. A few days later the child shows symptoms of cerebral dysfunction with further diarrhoea and vomiting and increased haemorrhagic diathesis swiftly followed by death (30th day). The chronological progression of this case shows a hiatus between the acute haemolysis on the one hand and the acute renal failure, haemorrhagic diathesis and hemiparesis on the other. There is however no doubt that increased haemolysis is a constant factor throughout the course of the disease.
The autopsy (p. no. 1258/54) indicates as an underlying condition thrombotic microangiopathy as described by Symmers with involvement of the small arteries, arterioles and capillaries in the myocardium, kidneys, brain, lungs, colon, thyroid gland and adrenal glands. Small, ischaemic necrotic lesions have developed in the myocardium and there is linear subcortical softening in the left hemisphere of the brain, which explains the right-sided hemiparesis. However, renal failure is also caused in this case by confluent, patchy renal cortical necrosis (Fig.3) There is also evidence of haemorrhagic diathesis with mild skin purpura, anaemia and splenic and renal haemosiderosis as well as haemorrhagic and ulcerative colitis.
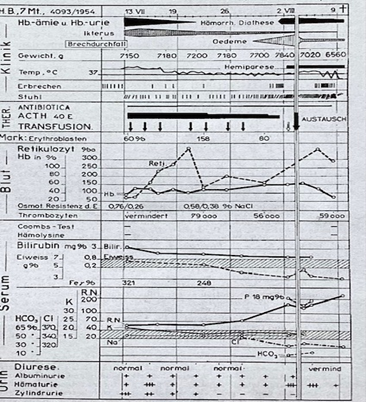
Fig 2*
Course curve Case 3
Case 4. T. Ruth, DOB 29 November 1952, J. No. 2553/54. 13-month-old girl suffering from acute otitis and severe fibrinous pleuropneumonia. High temperature on admission; no pronounced anaemia at this stage. Appearance of severe symptoms on the second day whilst receiving intensive penicillin treatment: oliguria (25 cm3), brownish-red urine with haemoglobinuria and severe haematuria. Transition to almost complete anuria. Haemoglobin levels fall, erythropoiesis remains inhibited, no evidence of autoantibodies. Reduced platelet count. Despite this, X-ray and clinical findings indicate an improvement in the pneumonia symptoms.
Because of vomiting a continuous infusion is set up, however the fluid requirement is overestimated, leading to significant weight gain and appearance of massive oedemas. Because of a further fall in haemoglobin levels, on 8 January an exchange transfusion is performed, as the blood chemistry also showed significant changes such as a further increase in BUN levels, hyperkalaemia and metabolic acidosis.
The existing hypoelectrolytaemia with reduced chlorine and sodium levels shows that the renal failure was partially extrarenal in origin. The exchange transfusion results in an improvement in the blood chemistry and initially stops the fall in haemoglobin levels. Potassium levels normalise, the hypoelectrolytaemia is resolved. Despite this, the oedemas continue to increase, there is an episode of severe diarrhoea and vomiting and finally a fall in haemoglobin levels, bleeding and spasms with death on the 12th day.
The anatomic pathology report (p. no. 102/54) indicates full-blown bilateral renal cortical necrosis ( Fig 3 ). Apart from a narrow subcortical area and a few linear bands, the cortical tissue is completely necrotic. The great vessels of the corticomedullary boundary are intact and freely accessible and only within the areas of infarction are the arterial walls affected by necrosis and their lumina thrombosed. In the upper and middle lobes of the right lung there is confluent and abscessing bronchopneumonia with concomitant serofibrinous pleuritis. Extensive secondary anaemic infarcts are found in the area affected by pneumonia, and according to the histology findings these are as old as the renal cortical necrosis and cannot in this case be attributed to a primary vascular disease or a thrombotic or embolic vascular occlusion. The autopsy also indicates uraemic gastroenteritis, with eosinophilia of the anterior pituitary gland of the endocrine system, epithelial and adrenal hyperplasia, cardiomegaly with myocardial lipidosis and acute organ congestion.
Case 5. G. Walter, DOB 27 January 1951, J. No. 7566/52. 14-month-old baby. Following acute fibrinous -purulent pleuropneumonia, there is increasing pallor with the development of almost complete anuria as well as tonic-clonic seizures with loss of consciousness. There is anaemia (haemoglobin 16%) with severe bone marrow erythroblastopaenia (0.6%) and very low blood reticulocyte count (1‰); repeated serology tests detect intensely avid autoantibodies (direct Coombs test ++) and the phenomenon of polyagglutinability. Moderate thrombocytopaenia, neutrophilic leucocytosis. Death occurs on the fourth day despite an exchange transfusion, antibiotics and ACTH as well as several blood transfusions.
The autopsy (p. no. 429/52) indicates strikingly similar findings to the previous case: bilateral renal cortical necrosis, only mild haemoglobinuria. Here too, extensive secondary anaemic infarcts with concomitant fibrinous-purulent pleuritis are found in the area affected by pseudo lobar, abscessing and haemorrhagic bronchopneumonia in the right upper lobe (Fig 3). In addition to haemorrhagic diathesis and anaemia there is acute organ congestion with cerebral oedema, dilatation of the left ventricle and of the right heart chambers.
Discussion of findings
Clinical picture
The clinical picture of all these fatal acute haemolytic-uraemic syndromes is extremely similar and consists of the following four main symptoms:
- acquired haemolytic anaemia,
- acute renal failure,
- haemorrhagic diathesis,
- cerebral symptoms.
- Acute acquired haemolytic anaemia is associated with intravascular haemolysis and
haemoglobinuria, morphologically severe erythrocyte changes and reduction in osmotic resistance. Autoantibodies (with the phenomenon of polyagglutinability) were evident only in case 5; however, the short lifespan of the patient’s own and transfused erythrocytes suggests that a haemolytically active substance is also present – albeit not detectable – in the plasma in the other cases. Erythropoietic regeneration is initially inhibited in most cases and in case 5 there is even severe erythroblastopaenia in the bone marrow (0.6%). In three cases haemolytic anaemia is triggered by torpid purulent airway infections; in case 3 infectious, bacteriologically unexplained enterocolitis is a possible cause and in case 1 there is no previous disease. In none of the cases is there evidence of exogenous noxious substances such as toxins or of erythropoiesis. The extent to which antibiotics and sulfonamide caused haemolytic activity and toxicity in case 2, and whether they led to Herxheimer reactions in cases 4 and 5, remains unclear, however. A further possible cause is absorption of abnormal proteins from tissue decay with resultant antigenic instability (cases 4 and 5).
- The second main symptom and one which is key to the deleterious nature of the disease is
acute renal failure with the following findings: oliguria or even complete anuria, constant albuminuria throughout the duration of the disease, i.e. not only in the initial phase of haemoglobinuria. Constant, at times massive haematuria, but only transient and less pronounced cylindruria. In case 1, specific gravity fluctuates between 1020 and 1014 (uncorrected in relation to albuminuria). In case 1, the oedemas are predominantly preorbital and already present at onset. In the cases where there is transition to anuria they increase and are at times very pronounced (in cases 4 and 5 probably also because of overly high fluid intake). In case 1, blood pressure is elevated from disease onset (not measured in the other cases [babies]). The blood chemistry changes resemble those of a complete shutdown of the kidneys.
The symptoms of kidney disorder in these cases must be distinguished from those of haemolytic-uraemic or crush syndrome, as far as the observations in adults (Swann and Merill) also apply to children; because of increasing oliguria that does not peak in the initial phase. The complete anuria observed in three cases and the specific gravity with values of over 1010 are not consistent with the clinical picture of crush syndrome. The periorbital oedemas observed at disease onset are more consistent with the clinical picture of acute glomerulonephritis. In terms of blood chemistry, a distinction is not possible, as both in these cases and in the oliguric phase of crush syndrome kidney function has virtually shut down: there is increasing retention of waste products (BUN and phosphorus) as well as metabolic acidosis. Potassium behaviour is dependent not only on insufficient renal excretion, but also on the intensity of tissue catabolism (occurring due to haemolysis or tissue decay), on pH and on as yet unknown factors affecting the cell membrane. Except for case 4 there is no evidence for the presence of azotaemia due to low fractional excretion of sodium, because there is no concomitant hyponatraemia. Hypochloraemia is probably the result of severe vomiting.
In addition to haemolytic-uraemic or crush syndrome other kidney diseases with concomitant anaemia require differential diagnosis. These are certainly not cases of blood loss anaemia, nor of acute erythroblastopaenia due to anuria (Richet, Alagille and Fournier), as haemolysis is not present in this. It is more difficult to distinguish these cases from acquired haemolytic anaemias in acute glomerulonephritis, of which we also observed several cases. In acute glomerulonephritis, however, acquired haemolytic anaemia is only a concomitant symptom and in any case neither precedes nor accompanies kidney changes. In literature severe kidney disorders associated with acquired haemolytic anaemias are seldom mentioned (Gasser, Wagner). There are known cases of severe kidney disorders in the sense of haemolytic-uraemic or crush syndrome caused by exogenously induced haemolysis (transfusion incident, intravenous administration of distilled water). There is clinical similarity with the symptoms of blackwater fever and certain forms of paroxysmal cold haemoglobinuria (Sussman and Kayden). Hepatorenal syndromes (leptospirosis, etc.) are excluded because of good liver function test results (normal urobilin quotient, normal prothrombin time, etc.).
- The third, less noticeable symptom is haemorrhagic diathesis with thrombocytopenic
purpura and bruising. It is interesting that in case 3, despite thrombocytopaenia, haemorrhagic diathesis is not initially clinically present. In case 1 there was evidence of significantly increased non-heparin-type serum antithrombin, which is also an indication of allergenic processes.
- The fourth symptom is psychological and neurological changes. In case 1 they are not very
noticeable (only euphoria); in cases 2, 4 and 5 there are severe cerebral spasms and in case 3 pronounced hemiparesis.
Our therapeutic approach has failed. It consisted mainly of ACTH, transfusions and exchange transfusions, antibiotics to treat infections as well as extensive renal protection. However, in such cases fluid intake must be better calculated; great caution is also indicated with blood transfusions. A new case has been treated with ACTH, cortisone and renal protection and the patient has almost fully recovered.
Anatomic pathology examination
In none of the five cases did the anatomic pathology examination find the cause of renal failure to be common haemolytic-uraemic or crush syndrome. However, because of the detection in clinical tests of haemoglobinaemia and haemoglobinuria, varying levels of haemoglobin casts are found in the collection tubes, and in the case with the longest disease progression (case 3) a low level of haemoglobin is also found in the renal tubular epithelial cells. Only in one case (case 1) is there evidence of tubular necrotic lesions characteristic of crush syndrome. The cause of fatal uraemia common to all five cases, however, is bilateral renal cortical necrosis, the extent of which, as can be seen in the diagram in Fig. 3, ranges from pure glomerular necrotic lesions to larger patchy necrotic lesions to complete cortical infarction. The location of the ischaemic necrotic lesions explains the different clinical symptoms. Thus, the symptoms in the first case were those of acute glomerulonephritis. Dunn and Montgomery have also described these changes as necrotising glomerulonephritis. Together with Blainey and based on the detailed investigations of Sheehan and Moore, we understand this to be a specific form of bilateral renal cortical necrosis.
Because of the longer, biphasic clinical progression of the disease, case 3 has particular significance. The autopsy in this case indicates thrombotic microangiopathy as described by Symmers as an underlying condition, which in addition to multiple other ischaemic necrotic lesions is complicated by bilateral renal cortical necrosis. This generalised vascular disease does not usually lead to such major ischaemic infarctions or to severe kidney damage. That said, Hunt has already reported a case of bilateral renal cortical necrosis with thrombocytopenic purpura, which certainly seems to correspond to our observation. The clinical picture of bilateral renal cortical necrosis, rare in itself, is most frequently – in approximately two-thirds of cases – observed in women at the end of pregnancy or following birth, in particular in late toxicosis or premature placental abruption. However, the extensive studies conducted by Sheehan and Moore as well as by Zuelzer & Co. show that it also occurs in adults and children in connection with a wide variety of infections and intoxications, which seem completely unconnected with each other.
| What is remarkable about our observations is that this severe kidney injury occurred at the same time as acute intravascular haemolysis or as a consequence of it. In a very thorough study of all the literature, with the exception of the above-mentioned case reported by Hunt, we have found only one single reference to bilateral renal cortical necrosis in a girl with haemolytic anaemia (Sheehan and Moore, case 69). The link between acute haemolysis and ischaemic kidney damage remains uncertain. What one can be certain of is that all forms of bilateral renal cortical necrosis are a result of functional ischaemia. Sheehan and Moore posit artery spasms that lead, depending on their location, to glomerular necrosis or to complete cortical necrosis. For Allen, cortical necrosis is no more than the most severe consequence of shock resulting from a complete blockage of the renal blood flow. |
Fig 3* Cases summary of disease course and diagnosis
This reduction in renal blood flow is also recognised by Zollinger as a significant factor in the development of haemolytic-uraemic syndrome. However, it should not lead either to isolated cortical ischaemia or to morphologically detectable ischaemic necrosis. From experimental studies it also appears unlikely that the released haemoglobin itself leads to ischaemic necrosis. It seems rather more likely that strong antigen antibody reactions occurred in these children following the severe and mild torpid infections that were evident in the majority of cases. On the one hand these would have led to massive intravascular haemolysis and on the other – perhaps with reflex vasospasms – to renal cortical necrosis and, as in the last two cases, to infarcts in the infectious/inflammatory foci.
However, we wish to stress here that acute renal failure observed in haemolytic processes can be caused not only by common crush syndrome, but also by such renal cortical necrosis.
Summary. In five children, four girls and one boy, aged two months, seven months, 13 months, 14 months and seven years, severe renal failure concomitant with or because of acute acquired haemolytic anaemia was observed that did not clinically correspond to the symptoms of haemolytic-uraemic or crush syndrome and despite all therapeutic efforts led to death within three to 12 days. Thrombocytopenic purpura and cerebral symptoms were also present. In the majority of cases haemolytic-uraemic syndrome occurred following a mild or severe infection and in two cases following toxic croupous pleuropneumonia. In all cases the autopsy surprisingly identified the cause of renal failure to be bilateral renal cortical necrosis and in one case thrombotic microangiopathy as described by Symmers as an underlying condition.
References:
Allen, A. C.: The Kidneys, Grune and Stratton, 1951.
Blainey, J. D.: J. Path. Bact. 64, 121 (1952).
Dunn, J. S., and Montgomery, G. L.: J. Path. Bact. 52, 1 (1941).
Evans, R. S., Takahashi, K., Duane, R. T., Payne, R., and Chi-Kong-Lin: Arch. intern. Med. 87, 48 (1951).
Gasser, C.: Die hämolytischen Syndrome im Kindesalter [Haemolytic syndromes in children]. Thieme, Stuttgart 1951.
Hunt, F. G.: J. roy. nav. med. Serv. 25, 270 (1939), quoted in Sheehan and Moore.
Moschcowitz, E.: Arch. intern. Med. 36, 89 (1925).
Oliver, J., McDowell, M., and Tracy, A.: J. clin. Invest, 30, 1305 (1951).
Richet, G., Alagille, D., and Fournier, E.: Presse méd. 62, 50 (1954).
Sheehan, H. L., and Moore, H. C.: Renal Cortical Contraction of the Kidney and Concealed Accidental hemorrhage. C. C. Thomas, Springfield, 1952.
Singer, K., Bornstein, F. P., and Wile, S. A.: Blood 2, 542 (1947).
Sussmann, R. M., and Kayden, H. J.: Arch. intern. Med. 82, 598 (1948), quoted in Oliver, McDowell and Tracy.
Swann, R. C., and Merill, J. P.: Medicine 32, 215 (1953).
Symmers, W. St. C.: Brit. med. J. 1952/2, 897; Verh. dtsch. Ges. Path. 36 Tg., P. 224. 1953.
Wagner, J.: Great Ormond Street J. 1954, No. 7, 66.
Zollinger, H. U.: Anurie bei Chromoproteinurie [Anuria in chromoproteinuria]. Thieme, Stuttgart 1952.
Zuelzer, W. W., Seymour, C., Kurnetz, R., Newton, W. A., and Fallon, R.: Amer. J. Dis. 81, 1 (1951).
Discussion:
R. E. Siebenmann (postscript): The pathogenesis of haemolytic-uraemic or crush syndrome is today explained as, on the one hand, the consequence of renal ischaemia with tubular necrosis (tubulorrhexis, Oliver), and on the other hand kidney injury (glomerulotubular nephropathy, Zollinger). Acute intravascular haemolysis very rarely leads to fatal kidney damage. These cases of renal cortical necrosis may be the consequence of severe shock. Morphological examination is unable to clarify whether this is thromboplastin shock.
Note: The original article was published in German in the Swiss Medical Weekly ( Schweizerische Medizinische Wochenschrift 38/39) in the week commencing 20th September 1955.
* Translation of terms in illustrations: acute erworbene hamolytisch anamie – acute acquired haemolytic anaemia, akut anuria mit uramie – acute anuria with uraemia, cerebrale krampte- cerebral cramps, antibiotica- antibiotics, albuminurie- albuminuria, austausch- exchange brechdurchfall- vomit, blut- blood, eiweiss- protein , erbrechen- vomit, erithroblasten- erythroblasts, gewicht- weight, hamorrhage diathese- bleeding tendency, hamatuie- haematuria, hamolysin – haemololysin, hemiplege – haemiplegia , hemiparese – haemoparesis, ikterus – jaundice, klinik- clinic, jahre – years, monate – months oedema – oedema, , retikulozyt – reticulocyte, stuhl- bowel movement, tage – days, thrombozyten- platelets, thromopenie – thrombopenia , thrombopenische purpura – thrombopenia purpura, terminal krapte terminale krampft – terminal spasms, urin -urine, velauf- course, vermind- reduce, zylindruie- Cylinduria.
Article No. 800

{kind=link}